CRISPR gene editing reveals new therapeutic approach for blood disorders
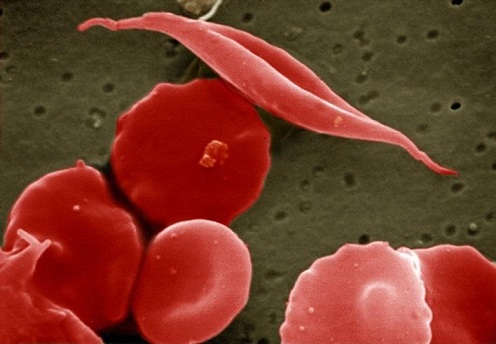
This is a scanning electron micrograph of sickled and other red blood cells.
An international team of scientists led by researchers at St. Jude Children's Research Hospital has found a way to use CRISPR gene editing to help fix sickle cell disease and beta-thalassemia in blood cells isolated from patients. The study, which appears online today in Nature Medicine, provides proof-of-principle for a new approach to treat common blood disorders by genome editing.
"Our approach to gene editing is informed by the known benefits of hereditary persistence of fetal hemoglobin," said Mitchell J. Weiss, M.D., Ph.D., chair of the St. Jude Department of Hematology and one of the study's lead authors. "It has been known for some time that individuals with genetic mutations that persistently elevate fetal hemoglobin are resistant to the symptoms of sickle cell disease and beta-thalassemia, genetic forms of severe anemia that are common in many regions of the world. We have found a way to use CRISPR gene editing to produce similar benefits."
Fetal and adult hemoglobin are two different molecular forms of the essential oxygen-carrying molecule in red blood cells. Hemoglobins are made up of different combinations of four molecular subunits. Sickle cell disease and beta-thalassemia are caused by mutations in a gene encoding an adult-expressed subunit termed "beta." Disease becomes apparent after birth as the levels of adult hemoglobin take hold and levels of fetal hemoglobin decline. These mutations can affect the survival of red blood cells and inhibit oxygen delivery to tissues, causing impaired function of different organs with devastating consequences for patients. Fetal hemoglobin lacks beta subunits and has gamma subunits instead. Thus, beta-thalassemia or sickle cell disease-associated mutations, which impair the production or function of the beta subunit, do not cause problems with fetal hemoglobin, which can transport oxygen effectively in adults.
Experts have known for some time that inhibiting or reversing "gamma-to-beta" switching of hemoglobin subunits can raise levels of fetal hemoglobin in adults and significantly ameliorate the debilitating symptoms of beta-thalassemia or sickle cell disease.
"Our work has identified a potential DNA target for genome editing-mediated therapy and offers proof-of-principle for a possible approach to treat sickle cell and beta-thalassemia," added Weiss. "We have been able to snip that DNA target using CRISPR, remove a short segment in a "control section" of DNA that stimulates gamma-to-beta switching, and join the ends back up to produce sustained elevation of fetal hemoglobin levels in adult red blood cells." When the scientists edited the DNA of blood-forming stem cells derived from patients with sickle cell disease, they were able to activate those genes and produce red blood cells that had enough fetal hemoglobin to be healthy.
Recently, scientists have used several gene editing approaches to manipulate blood-forming stem cells for the possible treatment of sickle cell disease and beta-thalassemia, including repair of specific disease-causing mutations and other strategies to inhibit gamma-to-beta switching. All of these approaches remain untested in patients.
"Our results represent an additional approach to these existing innovative strategies and compare favorably in terms of the levels of fetal hemoglobin that are produced by our experimental system," said Weiss. Using genome editing to restore the hereditary persistence of fetal hemoglobin is an attractive possibility, because it can be achieved relatively easily using current technologies. The condition is known to be benign in people who inherit similar naturally occurring mutations.
At this stage, the scientists emphasize that it is still too early to begin clinical trials of the new gene editing approach. The researchers want to refine further the gene editing process and perform other experiments to minimize potentially harmful off-target mutations before in-human clinical trials are considered. Additionally, it will be important to compare different approaches head-to-head to determine which one is safest and most effective.
Source: St. Jude Children's Research Hospital
BİOLOGY NEWS
-

RNA from Trained Aplysia Can Induce an Epigenetic Engram for Long-Term Sensitization in Untrained Aplysia.
-
HOW WE BECOME HAPPY
-
More tomatoes, faster: Accelerating tomato engineering
-
Purest yet liver-like cells generated from induced pluripotent stem cells
-
Legions of nanorobots target cancerous tumors with precision
-
CRISPR gene editing reveals new therapeutic approach for blood disorders
-
Quantum dots with impermeable shell: A powerful tool for nanoengineering
-
Thousands on one chip: New method to study proteins
-
Gene Drive Technology: Where is the future?
-
Four newly identified genes could improve rice
-
Watching the luminescent gene switch
-
DNA damage by ultrashort pulses of intense laser light
-
Mantis shrimp inspires next generation of ultra-strong materials
-
Specialized life forms abound at Arctic methane seeps
-
Ocean warming and acidification impact on calcareous phytoplankton
